
Informationen zur neuen EU Medizinprodukteverordnung 2017/745 (MDR)
Die folgenden Inhalte wurden von der pritidenta GmbH mit der größtmöglichen Sorgfalt erstellt.
Wir übernehmen keine Gewährleistung für die Aktualität, Richtigkeit und Vollständigkeit der auf dieser Webseite zur Verfügung gestellten Inhalte und schließen jegliche Haftung für Schäden aus, die durch die Nutzung der zur Verfügung gestellten Daten oder durch die Nutzung fehlerhafter oder unvollständiger Informationen verursacht wurde, sofern nicht unsererseits nachweislich ein vorsätzliches oder grob fahrlässiges Fehlverhalten vorliegt.
Was ist die MDR?
Die MDR (Medical Device Regulation) ist die neue europäische Medizinprodukte-Verordnung. Sie ist am 25. Mai 2017 in Kraft getreten und ab dem 26. Mai 2021 verpflichtend anzuwenden. Die Verordnung bildet den neuen EU-Rechtsrahmen für Medizinprodukte und löst die bisherige Medizinprodukte-Richtline MDD (Medical Device Directive (93/42/EWG)) ab.
Die MDR definiert erweiterte Pflichten für alle Wirtschaftsakteure (u.a. Hersteller und Händler), die Teil der Lieferkette eines Medizinprodukts sind. Die Erfüllung dieser Anforderungen ist die Voraussetzung um Medizinprodukte in Europa verkaufen zu dürfen.
Im Unterschied zur MDD muss die MDR als europäische Verordnung nicht erst in nationales Recht umgesetzt werden. Sie gilt daher ab dem 26. Mai 2021 unmittelbar in Deutschland.
Welche Ziele hat die MDR?
Die MDR soll
- Ein hohes Niveau an Gesundheitsschutz für Patienten und Anwendern gewährleisten
- Standards für die Erhöhung der Qualität und Sicherheit von Medizinprodukten festlegen
- Einen reibungslos funktionierenden EU-Binnenmarkt für Medizinprodukte garantieren
- Bestimmungen zur Gewährleistung von Transparenz und Rückverfolgbarkeit von Medizinprodukten über ihre gesamte Lebensdauer einführen
Umsetzungsfristen
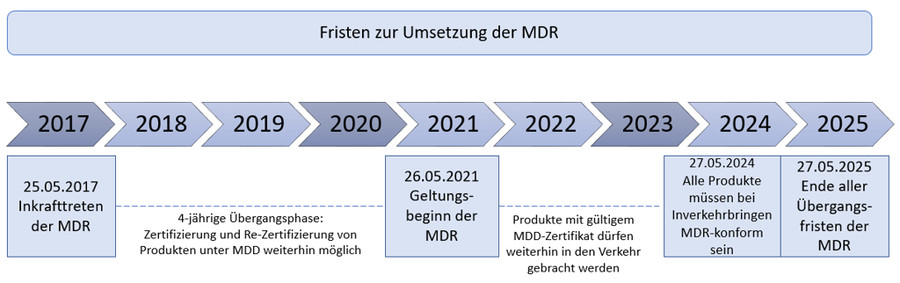
Wichtiger Hinweis: Alle Produkte, die bis zum 26. Mai 2024 unter der MDD in den Verkehr gebracht, dürfen bis zum 27. Mai 2025 weiterverkauft werden („Abverkaufsregel“).
► Händler dürfen MDD konforme Produkte, die bereits erstmalig in Verkehr gebracht wurden, längstens bis 27. Mai 2025 verkaufen. Produkte, die bis zum Ende dieser Übergangsfrist bereits erstmalig an Endnutzer (z.B. zahntechnische Labore) abgegeben wurden können dagegen von ihnen auch nach diesem Zeitpunkt weiter genutzt werden.
MDR: Was ist neu?
- Die MDR führt erstmal den Begriff des „Wirtschaftsakteurs“ (darunter fallen unter anderem Hersteller und Händler) ein und legt deren Pflichten fest.
- Die Anforderungen für die Hersteller an den Inhalt der Technischen Dokumentation werden deutlich umfangreicher. Die Unterlagen müssen kontinuierlich aktualisiert werden.
- Die Pflichten zur Marktbeobachtung nach dem Inverkehrbringen („Post-Market-Surveillance“) wurden erheblich erweitert. Davon sind nicht nur Hersteller, sondern auch die anderen Wirtschaftsakteure betroffen.
- Klinische Bewertungen und klinische Prüfungen werden hinsichtlich der Art und Qualität der klinischen Daten detaillierter geregelt und vorgeschrieben.
- Die Anforderungen an die Informationen, die mit einem Medizinprodukt mitgeliefert werden müssen (Kennzeichnung, Gebrauchsanweisung) wurden signifikant erweitert.
- Jedes Produkt muss zukünftig eine eindeutige Produktidentifizierungsnummer (UDI) erhalten.
- Alle Medizinprodukte müssen als Medizinprodukte gekennzeichnet werden. pritidenta wird mit dem Symbol „MD“ darauf hinweisen. Dieses Symbol drucken wir direkt auf die Verpackung.
- Die Klassifizierung einiger Produkte ändert sich.
- Medizinprodukte-Hersteller müssen eine qualifizierte Person im Unternehmen benennen, die umfangreiches Fachwissen auf dem Gebiet der Medizinprodukte besitzen muss und für die Einhaltung der Regulierungsvorschriften verantwortlich ist.
- Die Aufbewahrungsfrist für Dokumente wurde deutlich von 5 auf 10 (für implantierbare Produkte auf 15) Jahre verlängert.
- Die Anforderungen an die Benannten Stellen sind erhöht worden. Zugleich sind sie künftig verpflichtet, mindestens einmal alle 5 Jahre unangekündigte Audits bei Herstellern durchzuführen.
- Die europäische Datenbank EUDAMED wird deutlich ausgeweitet. Bisher konnten nur staatliche Institutionen darauf zugreifen. In Zukunft haben auch Hersteller, Benannte Stellen und die Öffentlichkeit auf sie Zugriff.
Werden die Produkte von pritidenta ab Mai 2021 weiter geliefert?
pritidenta hat die Erstzertifizierung unter der MDR im März 2022 abgeschlossen. Alle zertifizierten Produkte können somit über den Stichtag 27.05.2024 weiter In Verkehr gebracht werden.
Wie stellt pritidenta als Hersteller die Einhaltung der MDR sicher?
pritidenta hat sein Qualitätsmanagementsystem und Technische Dokumentation an die Anforderungen der MDR angepasst. Die Zertifizierung unserer Produkte nach MDR haben wir im März 2022 abgeschlossen. Das Zertifikat können Sie im Downloadbereich einsehen.
Unsere Benannten Stelle, die mdc medical certification GmbH, hat bereits am 25. April 2020 die offizielle Benennung unter der Verordnung (EU) 2017/745 (MDR) erhalten und erfüllt somit in vollem Umfang die Anforderungen der MDR.
Was verlangt die MDR von Ihnen als Händler (Händlerpflichten)?
Wenn Sie Kunde von pritidenta sind und unsere Produkte weiterverkaufen nehmen Sie immer die Rolle des Händlers ein. Unter der MDR entstehen somit auch neue Pflichten für Sie, auf die wir Sie gerne aufmerksam machen möchten.
Die folgenden Pflichten gelten für Sie, solange sich das Produkt in Ihrem Verantwortungsbereich befindet. Sollten Sie Grund zur Annahme haben, dass ein Produkt nicht den Anforderungen der MDR entspricht, dürfen Sie dieses Produkt nicht auf dem Markt bereitstellen.
Überprüfung des Produkts und der Dokumente vor der Bereitstellung auf dem Markt
- Sie überprüfen, ob das Produkt eine gültiges CE-Kennzeichnung hat. Sie finden das CE-Zeichen auf der Verpackung und auf dem Produkt selbst. Unsere Produkte gehören zur Risikoklasse IIb. Bei Medizinprodukten der Klassen IIa und höher muss neben dem CE-Zeichen auch immer die Kennnummer der Benannten Stelle stehen. Die Nummer unserer Benannten Stelle lautet 0483.
- Sie überprüfen, ob eine gültige EU-Konformitätserklärung für das Produkt in den Amtssprachen der EU-Mitgliedsstaaten, in denen Sie das Produkt verkaufen, vorliegt. Unsere Konformitätserklärung (zurzeit in Deutsch und Englisch) können Sie auf unserer Website herunterladen.
- Sie überprüfen, ob die Kennzeichnung und die Gebrauchsanweisung in der vom jeweiligen Land festgelegten oder akzeptierten Sprache, in dem das Produkt bereitgestellt werden soll, erstellt wurden. Bitte beachten Sie, dass pritidenta die Gebrauchsanweisungen für ihre Produkte grundsätzlich elektronisch auf ihrer Webseite zur Verfügung stellt. Die Gebrauchsanweisungen können Sie unter dem folgenden Link herunterladen: www.pritidenta.com/IFU.
- Sie überprüfen, ob auf dem Etikett der Verpackung eine UDI angegeben ist.
Diese Prüfpflichten können durch eine Stichprobenprüfung abgedeckt werden.
Sicherstellung der Lagerungs- und Transportbedingungen
- Sie stellen sicher, dass das Produkt entsprechend den festgelegten Vorgaben gelagert und transportiert wird. Diese Informationen können Sie jederzeit der Verpackung und der Gebrauchsanweisung entnehmen.
Mitwirkung bei der Marktüberwachung
- Sie sammeln Beschwerden und Berichte über Vorkommnisse, die im Zusammenhang mit unseren gelieferten Produkten entstanden sind und melden uns diese.
- Sie führen ein Register der Beschwerden, nicht-konformen Produkten und der Rückrufe bzw. Rücknahmen und halten es aktuell. Auf Anfrage stellen sie uns diese Informationen zur Verfügung.
- Bei Verdacht einer Produktfälschung oder einer schwerwiegenden Gefahr, die von unseren Produkten ausgeht, informieren sie unverzüglich die zuständigen Behörden.
- Sie arbeiten mit den Behörden zusammen und stellen alle gesammelten Informationen über das Produkt bereit.
Sicherstellung der Rückverfolgbarkeit
- Sie dokumentieren von wem Sie ein Produkt direkt bezogen und an wen Sie es direkt abgegeben haben.
- Sie bewahren diese Informationen für mindestens 10 Jahre (bzw. 15 Jahre bei implantierbaren Produkten wie den Produkten von pritidenta) auf und können sie auf Anfrage der Behörde zur Verfügung stellen.
Welche Informationen stellt pritidenta in Zusammenhang mit der MDR zur Verfügung?
Sie können u.a. folgende Informationen anfordern bzw. herunterladen:
Wo finden Sie weitere Informationen?
- Vollständiger Text der MDR: https://eur-lex.europa.eu/legal-content/DE/TXT/?uri=CELEX:32017R0745
- Informationen der EU-Kommission für Bevollmächtigte, Importeure und Händler: Authorised Representatives, Importers and Distributors | Public Health (europa.eu)
- Zugang zu den in EUDAMED gespeicherten Daten der Wirtschaftsakteure: https://ec.europa.eu/tools/eudamed/#/screen/search-eo
- Aktuelle Liste der Benannten Stellen, die unter der MDR zertifiziert sind: https://webgate.ec.europa.eu/single-market-compliance-space/home
Noch Fragen?
Wenn Sie Hilfe oder weitere Informationen benötigen – wir helfen Ihnen gerne weiter. Schicken Sie uns einfach Ihre Fragen an quality[at]pritidenta.com – wir melden uns umgehend bei Ihnen.
Verordnung
Die MDR ist eine EU-Verordnung. EU-Verordnungen (englisch „regulation“) gelten in den
Mitgliedsstaaten der EU unmittelbar und müssen nicht erst in nationales Recht (z.B. als Gesetz)
umgesetzt werden. Sie sind in allen ihren Teilen verbindlich, d.h. Anpassungen durch einzelne
Mitgliedsstaaten sind grundsätzlich nicht möglich. Verordnungen können allerdings auch einzelne
Artikel enthalten, die Anpassungen an nationales Recht zulassen. In Deutschland dient
das Medizinprodukterecht-Durchführungsgesetz (MPDG) zur Durchführung und Ergänzung der MDR.
Richtline
Die MDD ist eine EU-Richtlinie. EU-Richtlinien (englisch „directive“) sind im europäischen Recht nicht
unmittelbar wirksam, sondern müssen von den Mitgliedsstaaten der EU erst in nationales Recht
umgewandelt werden. Die MDD wurde z.B. in Deutschland durch das Medizinproduktegesetz (MPG)
umgesetzt.
Wirtschaftsakteur
Der Begriff „Wirtschaftsakteur“ (englisch: economic operator“) wurde mit der MDR neu eingeführt.
Wirtschaftakteure sind Einheiten innerhalb der Lieferkette eines Medizinprodukts. Die MDR kennt
folgende Wirtschaftsakteure:
- Hersteller
- Bevollmächtigter Vertreter
- Importeur
- Händler
- Inverkehrbringer von Systemen und Behandlungseinheiten
Die MDR definiert für jeden Wirtschaftsakteur bestimmte Verantwortlichkeiten und Verpflichtungen.
Die wesentlichen Pflichten für Hersteller sind in Artikel 10 der MDR geregelt. Artikel 14 MDR legt die
allgemeinen Pflichten für Händler fest.
Inverkehrbringen
Unter der MDR die erstmalige Bereitstellung (d.h. die entgeltliche oder unentgeltliche Abgabe eines Produkts zum Vertrieb, zum Verbrauch oder zur Verwendung im Rahmen einer gewerblichen Tätigkeit) im EU-Markt.
Beispiel: Ein europäischer Hersteller verkauft sein Produkt erstmalig an einen europäischen Händler. Der Hersteller bringt dadurch das Produkt in den Verkehr und stellt es gleichzeitig bereit. Wenn der Händler das Produkt weiterverkauft, stellt er es bereit.
Hersteller
Ein Hersteller vermarktet ein Produkt unter seinem Namen oder seiner Marke. Dabei macht es keinen Unterschied, ob er es selber herstellt oder ob er es herstellen lässt.
Händler
Ein Händler stellt ein Produkt auf dem Markt bereit, nachdem es vom Hersteller oder Importeur in Verkehr gebracht wurde. Die allgemeinen Pflichten von Händlern sind in Artikel 14 der MDR geregelt.
EUDAMED (European Database on Medical Devices)
Die europäische Datenbank für Medizinprodukte ist das von der Europäischen Kommission entwickelte IT-System zur Umsetzung der Verordnung (EU) 2017/745 über Medizinprodukte und der Verordnung (EU) 2017/746 über In-vitro-Diagnostika. EUDAMED soll die Transparenz und Koordination von Informationen über Medizinprodukte, die auf dem EU-Markt erhältlich sind, verbessern.
Das System soll als Registrierungssystem, als Kooperationssystem, als Meldesystem und als (teilweise auch für die Öffentlichkeit zugängliches) Verbreitungssystem fungieren und interoperabel sein.
Es besteht aus 6 Modulen (1. Registrierung von Wirtschaftsakteuren („Actor registration“), 2. Unique Device Identification (UDI), 3. Bescheinigungen (“Certificate”), 4. Klinische Prüfungen (“Clinical investigation”), 5. Marktüberwachung („Vigilance“), 6. Marktbeobachtung (“Market surveillance”), von denen bisher nur die Registrierung von Wirtschaftsakteuren freigeschaltet ist (seit dem 01. Dezember 2020).
Hersteller, Importeure und bevollmächtigte Vertreter müssen sich gemäß MDR in EUDAMED registrieren. Für Händler ist keine Registrierung vorgesehen.
Technische Dokumentation
Die Technische Dokumentation für Medizinprodukte umfasst alle Dokumente, die die Hersteller bereitstellen müssen, um ihre Medizinprodukte einer Konformitätsbewertung zu unterziehen und Medizinprodukte auf den Markt bringen zu dürfen. Aufbau und Inhalt sind in der MDR detailliert vorgegeben.
Kennzeichnung
Geschriebene, gedruckte oder grafisch dargestellte Informationen, die entweder auf dem Produkt selbst oder auf der Verpackung jeder Einheit oder auf der Verpackung mehrerer Produkte angebracht sind.
UDI („Unique Device Identification“):

Die UDI ist eine weltweit eindeutige Produktidentifizierungsnummer für Medizinprodukte. Sie muss in maschinenlesbarer Form (beispielsweise Barcode) und in Klarschrift auf dem Produkt oder seinen Verpackungen aufgebracht werden.
pritidenta bringt die UDI bereits auf den Etiketten ihrer Verpackungen an und erfüllt damit die neuen MDR-Anforderungen.
Beispiel für eine UDI auf der Verpackung von pritidenta
Klassifizierung
Medizinprodukte werden in Europa aufgrund ihrer Zweckbestimmung in 4 unterschiedliche Risikoklassen eingeteilt (I, IIa, IIb, III). Die Klassifizierungsregeln sind im Anhang VIII der MDR festgelegt.
Benannte Stelle
Benannte Stellen (englisch „notified bodies“) sind in der EU staatlich benannte und staatlich überwachte private Auditier- und Zertifizierstellen, die im Auftrag der Hersteller tätig werden, um deren Konformitätsbewertung für ihre Erzeugnisse zu begleiten und zu kontrollieren. Um auch unter der MDR tätig werden zu können müssen sich die Benannten Stellen für Medizinprodukte einem umfangreichen Überprüfungsverfahren unterziehen, bevor sie neu „benannt“ werden.
Vorkommnis
Ein Vorkommnis gemäß MDR bezeichnet eine Fehlfunktion oder Verschlechterung der Eigenschaften oder Leistung eines bereits auf dem Markt bereitgestellten Produkts, einschließlich Anwendungsfehlern aufgrund ergonomischer Merkmale, sowie eine Unzulänglichkeit der vom Hersteller bereitgestellten Informationen oder eine unerwünschte Nebenwirkung.
Symbol „MD“

REF 291 Rev 000