
Information on the new EU Medical Device Regulation 2017/745 (MDR)
The following contents have been created by pritidenta GmbH with the greatest possible care.
We do not assume any warranty for the topicality, correctness and completeness of the contents provided on this website and exclude any liability for damages caused by the use of the data provided or by the use of incorrect or incomplete information, unless there is evidence of willful misconduct or serious negligence on our part.
What is the MDR?
The MDR (Medical Device Regulation) is the new European medical device regulation. It entered into force on May 25, 2017, and its date of application is mandatory as of May 26, 2021. The regulation forms the new EU legal framework for medical devices and replaces the previous Medical Device Directive (MDD (93/42/EEC)).
The MDR defines extended obligations for all economic operators (including manufacturers and distributors) who are part of the supply chain of a medical device. Compliance with these requirements is a prerequisite for placing the medical devices on the Europe market.
In comparison to the MDD, the MDR as a European regulation does not have to be implemented into national law first. It will therefore apply directly in Germany from May 26, 2021.
What are the objectives of the MDR?
The MDR is intended to
- ensure a high level of health protection for patients and users
- set standards for increasing the quality and safety of medical devices
- guarantee a functioning EU internal market for medical devices
- introduce provisions to ensure transparency and traceability of medical devices throughout their life cycle
Implementation deadlines
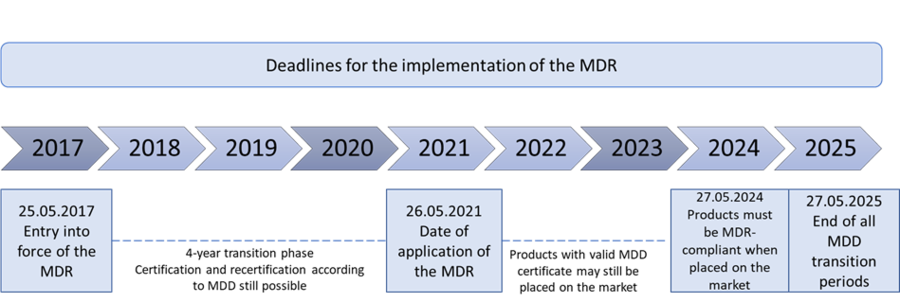
Important note: All products placed on the market under the MDD by May 26, 2024 may be continued to be distributed until May 27, 2025 ("sell-off clause").
► Distributors may sell MDD-compliant products that have already been placed on the market for the first time until May 27, 2025. Products that have already been supplied to end users (e.g. dental laboratories) by the end of this transitional period can be used after this date.
MDR: What's new?
- The MDR introduces the term "economic operator" (which includes, among others, manufacturers and distributors) and defines their obligations.
- The requirements for manufacturers with regard to the content of the Technical Documentation are significantly more extensive. Documents must be continuously updated.
- The obligations for post-market surveillance have been significantly expanded. This affects not only manufacturers, but also other economic operator.
- Clinical evaluations and clinical trials are regulated and required in more detail with regard to the form and quality of clinical data.
- The requirements for the information that must be supplied with a medical device (labeling, instructions for use) have been significantly expanded./li>
- Each product must be given a unique device identification number UDI in future.
- All medical devices must be labeled as medical devices. pritidenta will indicate this by using the "MD" symbol. We print this symbol directly on the packaging.
- The classification of certain products will change.
- Medical device manufacturers must designate a qualified person in the company who must have extensive expertise in the field of medical devices and is responsible for regulatory compliance.
- The document retention period has been significantly extended from 5 to 10 (for implantable devices to 15) years.
- The requirements for Notified bodies have been increased. At the same time, they will in future be obliged to perform unannounced audits of manufacturers at least once every 5 years.
- The European database EUDAMED has been expanded. Up to now, only government institutions had access to EUDAMED. In the future, manufacturers, Notified Bodies and the public will also have access to it.
Will pritidenta's products continue to be available as of May 2021?
pritidenta has completed the initial certification under the MDR in March 2022. All certified products can therefore continue to be placed on the market after the deadline of 27.05.2024
How does pritidenta as a manufacturer ensure compliance with the MDR?
pritidenta has adapted its quality management system and technical documentation to the requirements of the MDR. We have completed the certification of our products according to MDR in March 2022. You can view the certificate in the download area.
Our Notified body, mdc medical certification GmbH, has already received the official designation under Regulation (EU) 2017/745 (MDR) on April 25, 2020 and therefore fully complies with the requirements of the MDR.
What does the MDR require from you as a distributor distributor obligations)?
If you are a customer of pritidenta and sell our products, you always assume the role of a distributor. Under the MDR new obligations will arise for you, about which we would like to inform you.
The following obligations apply to you as long as the product is in your area of responsibility. If you have reason to believe that a product does not meet the requirements of the MDR, you are not allowed to make this product available on the market.
Examination of the product and the documents before making them available on the market
- You examine if the product has a valid CE marking. You can find the CE marking on the packaging and on the product itself. Our products are classified as risk class IIb. For medical devices of class IIa and higher, the CE mark must always be accompanied by the identification number of the Notified Body. The number of our Notified body is 0483.
- You examine if a valid EU Declaration of conformity for the product is available in the official languages of the EU member states in which you make the product available. You can download our Declaration of conformity (currently in English and German) from our website.
- You examine if the labeling uand the instructions for use have been prepared in the language specified or accepted by the country in which the product is to be provided. Please note that pritidenta generally provides the instructions for use for its products electronically on its website. You can download the instructions for use from the following link: www.pritidenta.com/IFU.
- You examine whether a UDI is stated on the label of the packaging.
These inspection obligations can be covered by a sample inspection.
Ensuring the storage and transport conditions
- You ensure that the product is stored and transported in accordance with the specified requirements. You can obtain this information at any time from the packaging and the instructions for use.
Participation in market surveillance
- You will collect complaints and reports of incidents that have occurred in connection with our supplied products and report them to us.
- You keep a register of complaints, non-conforming products and recalls or withdrawals and keep it up to date. Upon request, you will make this information available to us.
- In the event you suspect that the product is a falsified device or poses a serious risk, you shall immediately inform the relevant authorities.
- You will cooperate with the authorities and provide all collected information about the product.
Ensuring traceability
- You document from whom you have directly received a product and to whom you have directly supplied it.
- You keep this information for at least 10 years (or 15 years for implantable products such as pritidenta's products) and are able to make it available to the authorities upon request.
What information does pritidenta provide relating to the MDR?
You can request or download the following information:
Where can you find more information?
- Full text of the MDR Authorised Representatives, Importers and Distributors | Public Health (europa.eu)
- Access to economic operators' data stored in EUDAMED https://ec.europa.eu/tools/eudamed/#/screen/search-eo
- Current list of Notified Bodies certified under the MDR https://webgate.ec.europa.eu/single-market-compliance-space/home
Any questions?
If you need help or more information - we will be pleased to help you. Just send your questions to quality[at]pritidenta.com - we will contact you as soon as possible.
Regulation
The MDR is an EU regulation. EU regulations apply directly in the
member states of the EU and do not first have to be transposed into national law.
They are binding in all their parts. Adjustments by individual member states are generally not possible.
However, regulations can also contain individual articles that permit adaptations to national law.
In Germany, the Medical Device Law Implementation Act (MPDG) serves to implement and complement the MDR.
Directive
The MDD is an EU directive. EU directives are not directly effective in European law, but must
first be converted into national law by the EU member states. In Germany, for example, the
MDD was implemented by the Medical Devices Act (MPG).
Economic operator
The term "economic operator" was newly added to the MDR. Economic operators are
entities within the supply chain of a medical device. The MDR recognizes the following
economic operators:
- Manufacturer
- Authorized representative
- Importer
- Distributor
- Distributor of systems or procedure packs
The MDR defines certain responsibilities and obligations for each economic operator.
The main obligations for manufacturers are regulated in Artikel 10 der MDR of the MDR. Artikel 14 MDR MDR defines the general obligations for distributors.
Placing on the market
According to the MDR, the first time a product is made available (i.e., supplied against payment or free of charge for distribution, consumption or use in the course of a commercial activity) on the EU market.
Example: A European manufacturer sells his product to a European distributor for the first time. The manufacturer thereby places the product on the market and at the same time makes it available. When the distributor resells the product, he makes it available.
Manufacturer
A manufacturer markets a product under his name or brand. It makes no difference whether he manufactures it himself or has it manufactured.
Distributor
A distributor makes a product available on the market after it has been placed on the market by the manufacturer oder Importeur in Verkehr or importer. The general obligations of distributors are regulated in Artikel 14 of the MDR.
EUDAMED (European Database on Medical Devices)
The European Database on Medical Devices is the IT-system developed by the European Commission to implement Regulation (EU) 2017/745 on medical devices and Regulation (EU) 2017/746 on in vitro diagnostic medical devices. EUDAMED is intended to improve the transparency and coordination of information on medical devices available in the EU market.
The system is intended to function as a registration system, a cooperation system, a reporting system, a dissemination system (partially accessible to the public) and to be interoperable.
It consists of 6 modules (1. Actor registration, 2. Unique Device Identification (UDI), 3. Certificate, 4. Clinical investigation, 5. Vigilance, 6. Market surveillance), of which only the Actor registration has been activated so far (since December 01, 2020).
Manufacturers, importers and authorized representatives have to register in EUDAMED according to MDR. No registration is foreseen for distributors.
Technical Documentation
The Technical documentation for medical devices includes all documents that manufacturers must provide in order to have their medical devices subjected to a conformity assessment and to be allowed to place medical devices on the market. The structure and content are specified in detail in the MDR.
Labeling
Written, printed, or graphically represented information placed either on the product itself or on the packaging of each unit or on the packaging of multiple products.
UDI („Unique Device Identification“):

The UDI is a globally unique product identification number for medical devices. It must be applied to the product or its packaging in machine-readable form (e.g. barcode) and in plain text.
pritidenta already applies the UDI to the labels of its packaging and thus fulfills the new MDR requirements.
Example of an UDI on pritidenta's packaging
Classification
Medical devices in Europe are classified into 4 different risk classes (I, IIa, IIb, III) based on their intended purpose. The classification rules are defined in Annex VIII of the MDR.
Notified body
Notified bodies in the EU are government-appointed and government-monitored private auditing and certification bodies that act on behalf of manufacturers to accompany and monitor their conformity assessment for their products. In order to be able to operate under the MDR, the Notified Bodies for medical devices must pass an extensive inspection procedure before they are "re-designated".
Incident
An incident according to the MDR means a malfunction or deterioration in the characteristics or performance of a device already made available on the market, including use-errors due to ergonomic features, as well as an inadequacy of the information provided by the manufacturer or an undesirable side-effect.
Symbol „MD“

REF 292 Rev 000